
Fractal Therapeutics specializes in leveraging focused model-based analyses to advance drug R&D projects in a rigorous and efficient manner, without cutting corners. Learn more
Modeling should be performed in service of drug discovery and development, not the other way around. Our models deliver rigorous answers for practical questions in drug R&D. Learn more
There is hidden complexity in drug development - many choices are easy to make, but hard to get right. We help you make these choices in a data-driven way with fit-for-purpose models. Learn more

Fractal Therapeutics offers model-based drug discovery and development services that help make drug R&D more efficient. We help our clients answer the critical questions that the FDA (and investors) zero in on as they evaluate a molecule for its potential to help patients.
Our approach uses mathematical modeling, along with cost-efficient animal studies and focused clinical trial designs, to provide scientific input that is rigorous and timely.
Our team has spent many years in Big Pharma, learning what works and what doesn’t in drug development. We specialize in engaging with early-stage biotechs and academic groups, who often work with limited budgets and tight timelines. Our services help our clients build value all the way from project inception (Target Product Profile definition) to clinical proof-of-concept. We are drug development scientists, skilled at using model-based approaches to make better drugs, faster.
Our team members -having spent the past two decades working on drug discovery and development projects- have a broad skillset spanning many practical aspects of advancing drugs to the clinic (preclinical pharmacology, biomarker development, translational R&D and clinical trial design). Throughout this, a particular focus has been on using model-based analyses to quickly and efficiently advance drug discovery and development projects. Our parsimonious approach drives efficient project advancement while being underpinned by strong hypothesis-driven science.
While most pharma R&D projects are confidential, in some cases we have published the work or presented at major conferences. You can find a list of our publications on modeling and on drug discovery here and here, and a list of posters presented at major conferences here. Reprints are available on request.
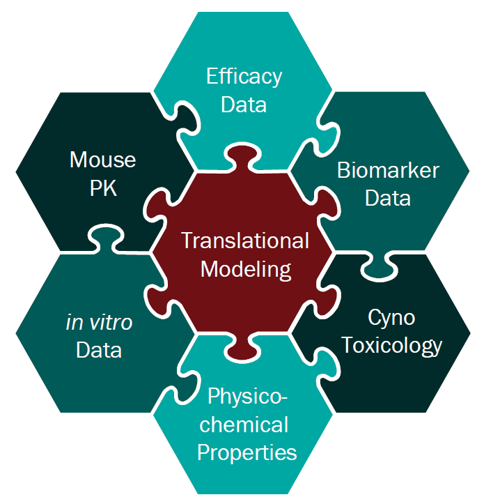
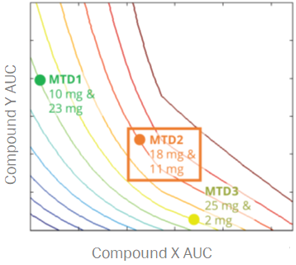
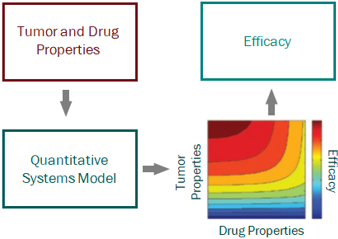
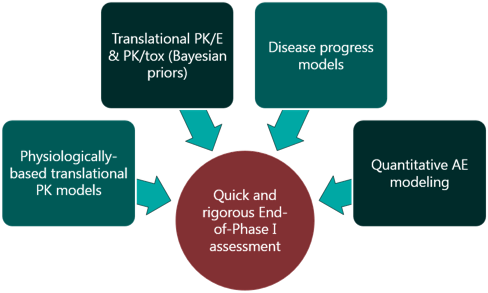

For a project that’s just starting out, our modeling can help define the whitespace for the Target Product Profile and inform the competitive strategy. Using publicly available or in-house data, we help clients answer the question “what makes a good drug for this project” and design an efficient screening strategy.

PK/PD and systems modeling are core competencies of ours. We use model-based drug development approaches, representing the cutting edge of big Pharma drug discovery and development practice, to provide guidance all the way from project inception to clinical proof of concept.

In vivo pharmacology and toxicology is where the rubber meets the road for a drug development program. These expensive experiments are critical for moving a program forward, and mistakes can seriously derail timelines and budgets, especially when using a CRO. Our deep background in in vivo pharmacology (both traditional and model-based) makes us the ideal partner for clients looking for a cost-effective, rigorous path to the clinic. We help clients get it right the first time!

For Oncology and Rare Diseases especially, focused yet rigorous biomarker strategies are the key to efficiently advancing molecules to and through early clinical development. Mathematical modeling plays a critical role in this, informing the choice of biomarkers, design of biomarker sampling schedules and supporting data interpretation every step along the way. Our team draws on years of experience in this space to help clients get their translational strategy right.

Clinical trial design can make or break a program- or company. There are many decisions that need to be made, and the traditional methods can perform surprisingly poorly in many cases. We help clients design cutting-edge adaptive trials, supported by rigorous statistical and mathematical modeling approaches, to enable efficient establishment of clinical POC

The pharmacology and toxicology sections of the IND are crucial pieces of the regulatory package, as they support the design choices made for First-in-Human trials and speak to the clinical potential of a molecule. We can provide you with the completed supporting package for these sections, including technical reports written to the FDA’s standards, as well as the IND section itself, and representation at regulatory meetings, if requested.
During the ongoing COVID-19 pandemic, we at Fractal Therapeutics have engaged on the emerging science and sought to contribute our modeling expertise in building a clearer understanding of the public-health risks.
We have worked as part of an interdisciplinary collaborative team to take a unique integrative approach tying together evolutionary biology, epidemiology and quantitative pharmacology with other engineering and science disciplines.
From very early on in the pandemic, we identified three major challenges:
We predicted (09/2020) that school reopening would lead to widespread disease propagation, which it did, and we showed that the CDC’s metrics for assessing disease spread in school are deeply flawed. Before the CDC announced that the “pandemic was over for the vaccinated”, we predicted (05/2021) that relaxing restrictions prematurely would lead to a variant-driven rebound in cases. That rebound was the Delta variant wave, in the fall of 2021.
Our collaborative work has proposed a mechanistic hypothesis for disease progression and has pointed out the critical importance of slowing the evolution of the virus during long-term infections. Our work predicts that future waves of the virus may lead to unpredictable (and potentially catastrophic) mortality burdens, and that vaccines alone cannot slow the evolution of this virus at this point. The current strategy of permitting widespread disease transmission while relying on immune responses alone to limit mortality is likely to be a fragile one. (This point has been picked up by Fortune magazine in a series of articles where they quote our work).
Going forward, we believe that there are several capabilities that governments and private enterprise must develop in order to bring this crisis to an end:
Our goal with working on COVID is to help flag the risks of the current strategy, and to contribute to the development of solutions to the crisis. It is our belief that the pandemic will not resolve itself without further biomedical and public-health intervention, and that attempting a return to pre-pandemic conditions without building new capabilities to slow the spread and evolution of this virus is recklessly optimistic.
(Click here for a full list of preprints, abstracts and papers by us on the topic of COVID-19)
By Ryan Nolan
"When I use a word,” Humpty Dumpty said, in rather a scornful tone, “it means just what I choose it to mean—neither more nor less.” “The question is,” said Alice, “whether you can make words mean so many different things.”
Lewis Carroll (1832–98)
It's a truism in pharma that a discipline is growing when no one can agree on what its basic terms mean anymore. The last ten years have seen a dramatic increase in the integration of modeling within the pharma. Many larger companies have one or more departments dedicated to the field, comprised of people with diverse backgrounds ranging from physics to engineering to pharmaceutical sciences. As a consequence of this melting pot of disciplines, modeling in the industry has seen its basic vocabulary blur in meaning. Ask a half-dozen different modelers to define the basic terms associated with PKPD modeling and you will get a dozen different answers. More importantly, if no one agrees on what the terms mean, then no one can be sure they are talking about the same things. As a result of such inconsistencies, it can be challenging to know the perspective and philosophy of a given modeler or modeling group. One step towards aligning expectations for modeling work is to understand the ambiguity in certain modeling concepts, and the back-story behind that ambiguity.
PKPD models
Despite being the most basic and fundamental term of all pharmaceutical modeling, the phrase PKPD is actually quite ambiguous. It’s not the fault of the PK part – most people are aligned on what constitutes pharmacokinetics – but rather it’s the PD that shoulders all the blame. Pharmacodynamics. Is it a biomarker measurement? Is it an efficacy readout? Is it a toxicity readout? Does it represent target engagement? How about pathway modulation? The short, yet unsatisfying answer, is yes…to all questions. PD can be any response that results from the drug, which is why PKPD is used so often, and also why it always requires further explanation. One improvement in such nomenclature is to specify the actual PD measurement, for example PK-E for efficacy, PK-Tox for toxicity, etc. I personally follow this approach and use PD for pathway biomarker measurement. In this manner, one can establish PK-RO-PD-E linkages (where RO is receptor occupancy). The take home here is if PD isn’t specified, make sure to ask what it represents.
Physiological models
Often known as Physiologically-Based PK models or PBPK models. These models have an interesting history. Their first use a few decades ago was to expand the application of a basic compartmental model, specifically to understand what the drug was doing at a specific tissue (i.e. site-of-action); an additional compartment representing a “realistic” tissue volume and flow to this tissue were incorporated. Over the next decade, as laboratory experiments further refined the physiological parameters (such as tissue volumes, blood flows, etc. of many species), the concept of adding more tissues and sub-compartments to these tissues became en vouge. The expectation was that the more “physiological” the model parameters, the more accurate it should be in representing the “true” system. Makes sense… except for one universal principle - all models are just an abstraction of reality. Using “real” parameters might make us feel warm and cozy, but at the end of the day, they are no more representative of the system than those from a one-compartment model. That’s not to say PBPK models aren’t useful; they certainly are, but the key is understanding what they really are – another, not more accurate, way of representing a drug in the body. Let’s get back to the timeline. During the mid-2000s PBPK models reached their peak of complexity with the application toward antibodies. The tissue compartments were divided into even more sub-sections to allow for endosomal compartment interactions and FcRn recycling kinetics. Over the next several years, these models were tested, until (most) people realized that obtaining measurements for such parameters is experimentally infeasible, and thus adding such extra complexity, while nice for academic publications, was not practically useful for drug development. In recent years everything has come full circle, with “physiological” models being applied, with actual utility, in the same manner as they were initially conceived, i.e. adding one or two extra compartments for tissues of interest – and of course given a new name (as if newly conceived) of “mini-PBPK” models.
Systems models
Hands down, the term “systems” model is the most ambiguous and widely misused term in the modern modeling world. The term Quantitative Systems Pharmacology, or QSP, models is used frequently, and has created an aura of impressiveness (mainly because no one really knows what it means or entails, but it sure sounds good). Let’s take a step back, again, a handful of decades. The term “systems” originated in traditional engineering disciplines as a means of describing a model with multiple interacting components; the components are somewhat independent sub-sets of the overall system, and are sometimes modular, and sometimes exist in different scales (in size and/or time). A systems model would, therefore, provide a means of integrating these separate components through mathematics to describe the behavior of the overall system. Around the 1970s systems approaches began to be applied to biological contexts, but it wasn’t until the 1990s, with the generation of genomic data, that the term “systems biology” began to be used more widely. At the turn of the century, systems biology modeling became hot as academics attempted to model multi-scale interactions among genes, proteins, metabolites, etc. across organ, tissue, and cellular levels. In such cases, application to pharmaceutical drug development was still quite limited. It wasn’t until the following decade that such modeling began to be incorporated into a pharmacology setting, with the interest of capturing all actions of a drug from target engagement through pathway signaling to cellular trafficking to disease modulation…and thus QSP was born. As with most grandiose construction, the concept is sometimes better than the product. In general, systems pharmacology models have their place in drug discovery and development – they are extremely useful when hypothesis testing mechanisms or integrating data from different experimental systems, but almost always in a fit-for-purpose, minimal structure. Where trouble can be had is when large (i.e. lots of parameters) models are constructed from multiple literature sources and pieced together to form a “definitive” model. We will speak to QSP models in more detail in a future post, but for now, the take home from systems models is this – be clear on what they entail, focus on only what can be measured, and stick to the mantra of less is actually more.
There are probably dozens of additional modeling terms that similarly get confused among industry scientists. Hopefully, these few examples are helpful in understanding why the ambiguity in terms can exist, and highlights the need for explicit communication regarding the modeling tools we are using, including their assumptions, limitations, and applications.
